Network Medicine Institute and Global Alliance
Representing 33 leading universities and institutions around the world committed to improving global health and advancing the field of Network Medicine

What is network medicine?
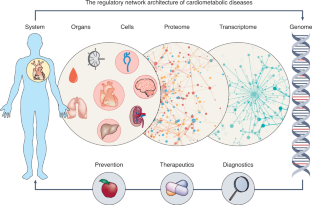
Network Medicine combines principles and approaches from network sciences, systems biology, and human dynamics to understand the causes of human diseases and develop new treatments.
History of Network Medicine
Network Medicine evolved from network science research during the early days of the internet and advances in systems biology since the human genome project. Now it's an established way to study, reclassify, and develop treatments for complex diseases.
1991
CERN introduces the World Wide Web to the public.
1999
Barabasi introduced the concept of scale-free networks and proposed the Barabási–Albert model to explain their widespread emergence in natural, technological and social systems. Barabasi’s paper on Scale-Free Networks in Science Magazine becomes the most-cited paper in the Physical Sciences.
2003
The Human Genome Project was declared complete in April 2003. The project determined there are approximately 22,300 protein-coding genes in human beings. Loscalzo and Barabasi begin building the map of human disease biology that explains how proteins expressed from the human genome interact to cause specific diseases.
2007
Loscalzo, Kohane, and Barabási publish Human disease classification in the postgenomic era in the Molecular Systems Biology journal which establishes Network Medicine as a complex systems approach to human pathobiology.
2011
Barabási, Gulbahce, and Loscalzo publish Network Medicine: A Network-based Approach to Human Disease in Nature where they present an overview of the organizing principles that govern cellular networks and the implications of these principles for understanding disease.
2012
The Channing Division of Network Medicine at Brigham and Women's Hospital was created to study, reclassify, and develop treatments for complex diseases using network science and systems biology.
2016
Joseph Loscalzo and Enrico Petrillo form the Network Medicine Alliance representing 31 leading universities and institutions around the world.
2017
Joseph Loscalzo, Albert-László Barabási, and Edwin Silverman publish the seminal Network Medicine textbook.
2018
Joseph Loscalzo and Albert-László Barabási publish Network-based approach to prediction and population-based validation of in silico drug repurposing which demonstrated that a unique integration of protein-protein interaction network proximity and large-scale patient-level longitudinal data complemented by mechanistic in vitro studies can facilitate drug repurposing.
The Transformation of Medicine, The First International Conference on Network Medicine and Big Data was held in Rome, Italy. The ultimate goal of the meeting was to design a strategy by which this interdisciplinary field can truly transform medicine.
2019
The Channing Division of Network Medicine (CDNM) continues to expand its staff of more than 80 Harvard Medical School faculty and 42 fellows in addition to 160 non-faculty Brigham and Women’s Hospital (BWH) employees. BWH oversees the second largest hospital-based research program in the world. CDNM’s annual research expenditures represented 25% of the BWH Department of Medicine annual budget. In fiscal year 2019, CDNM investigators received 54 new funding awards resulting in 173 active grants.
2020
In response to the COVID-19 pandemic, the Network Medicine Institute launches the COVID-19 Global Drug Repurposing Study (GDR Study). The GDR Study is based on a multifaceted drug repurposing strategy which includes a global patient registry and an advanced Network Medicine framework to identify promising therapies among the thousands of already FDA approved drugs. The strategy also facilitates evidence-based updates to health policy and clinical guidelines and tailors them to specific patient populations.
Selected Major Network Medicine Alliance consortium Grants 2022:

REPO4EU was funded with a highly competitive 5-year Twenty-Five Million Euro grant from the European Union Governments Horizons Research Program.
It is developing a comprehensive European and Global platform for precision drug repurposing applying existing drugs to be repurposed for diseases with significant unmet needs via innovative treatment protocols. The core ofREPO4EU consists of a team of world-leading scientists with breakthroughs in advanced bioinformatics and artificial intelligence (AI) on real-world big data to redefine diseases in a mechanism-based manner. This revolutionary new era of medicine will allow unprecedented efficacy and cost- effectiveness in treating diseases globally. Within 5 years, REPO4EU will establish a first-in-class coherent and innovative web-based platform for safe and efficient drug repurposing for all types of high unmet medical need indications to all European researchers and SMEs with a unique Open Science concept, ensuring global medical impact.
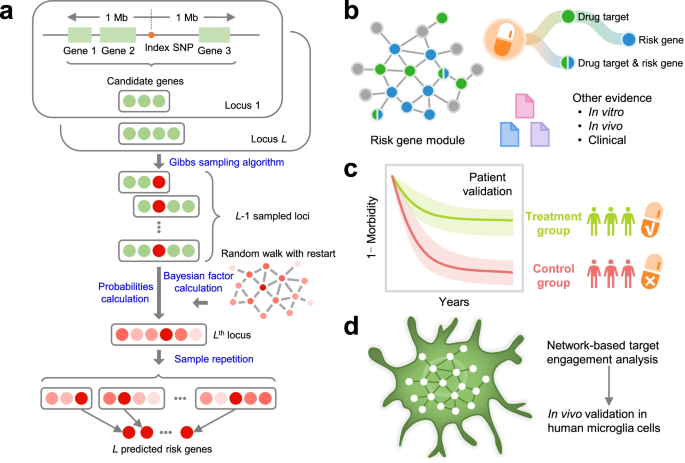
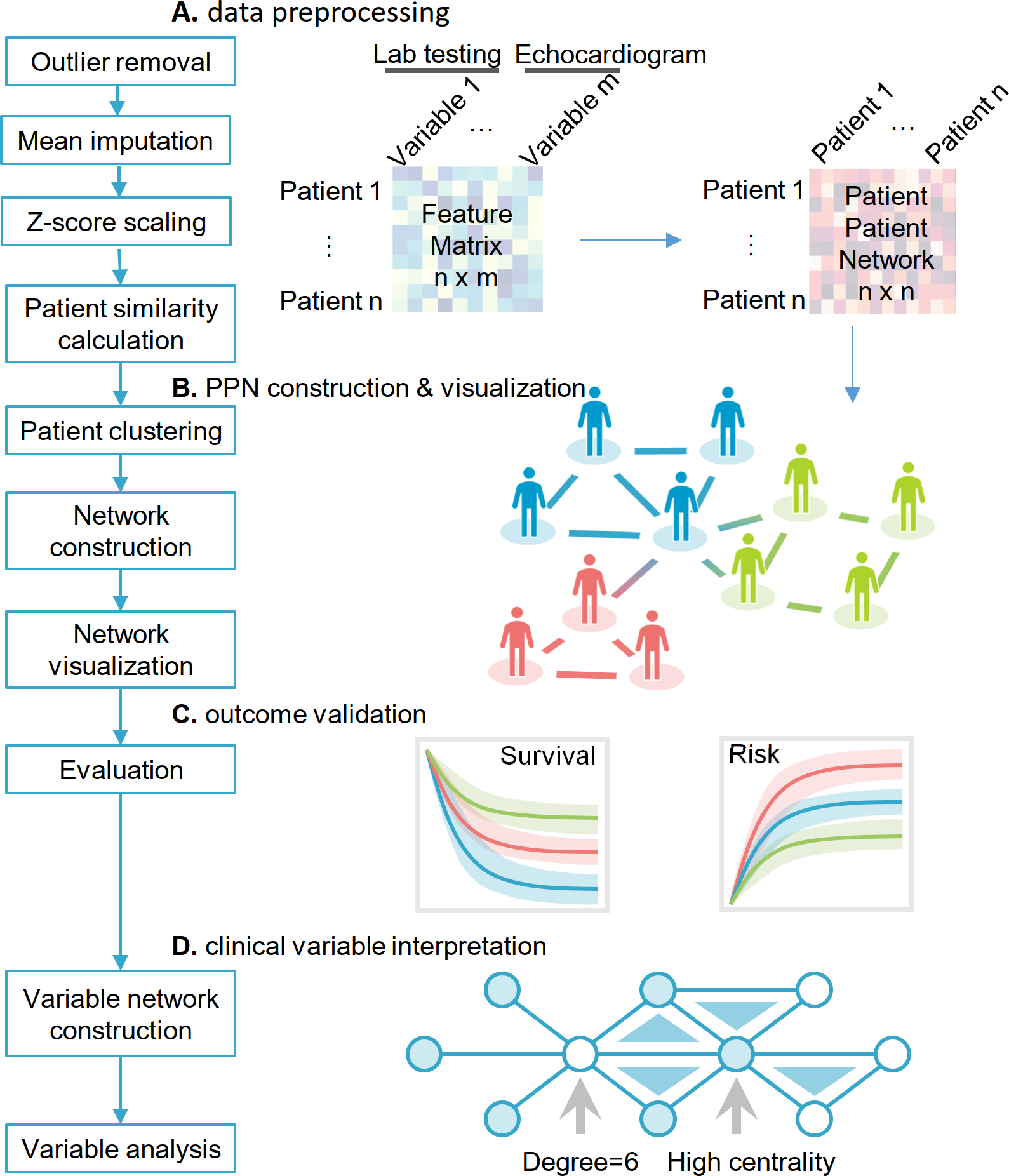
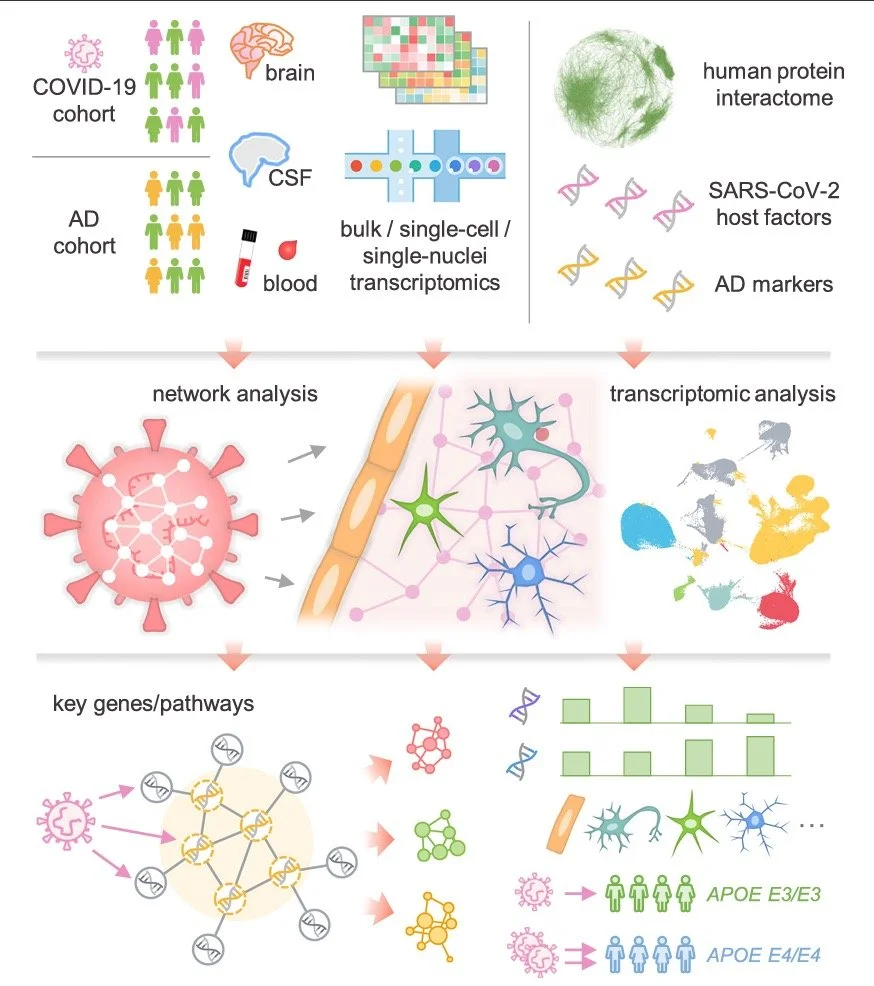
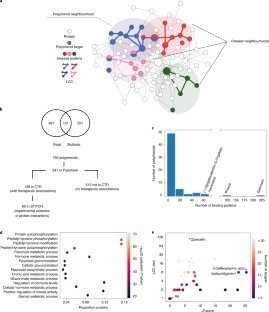
2021-2022 Research Highlights





New Members to the Network Medicine Alliance in 2021-2022
Medical University of Vienna (MedUni Vienna) is one of the most traditional medical education and research facilities in Europe. With almost 8,000 students, it is currently the largest medical training center in German-speaking countries. With 6,000 employees, 30 departments and two clinical institutes, 13 medical theory centers, and numerous highly specialized laboratories, it is also one of Europe's leading research establishments in the biomedical sector.
Paris-Saclay University is a public research university based in Paris, France. It is one of the 13 prestigious universities that were produced from the division of the University of Paris, known as the Sorbonne.
The Max Perutz Labs are dedicated to a mechanistic understanding of fundamental biomedical processes. By analyzing and reconstituting complex biological systems across different scales, our scientists aim to link breakthroughs in basic research to advances in human health.
The Complexity Science Hub Vienna is a Vienna-based research organization with the aim to bundle, coordinate and advance the research of complex systems, system analysis and big data science in Austria.
The University of Padua is one of Europe’s oldest and most prestigious seats of learning. As a multi-disciplinary institute of higher education, the University aims to provide its students with professional training and a solid cultural background. The qualification received from the University of Padua act as a symbol of the ambitious objectives respected and coveted by both students and employers alike.
Cyprus Institute of Neurology & Genetics
The Cyprus Institute of Neurology & Genetics (CING) is a private, non-profit, bi-communal, medical, research and academic center.
The INSTITUTE
The Institute's mission is to provide resources and guidance to alliance members so their research can have a global impact on health policy and clinical guidelines and untap the full potential of personalized medicine.
The Institute is a nonprofit with entities recognized by the United States and European Union.

THe Alliance
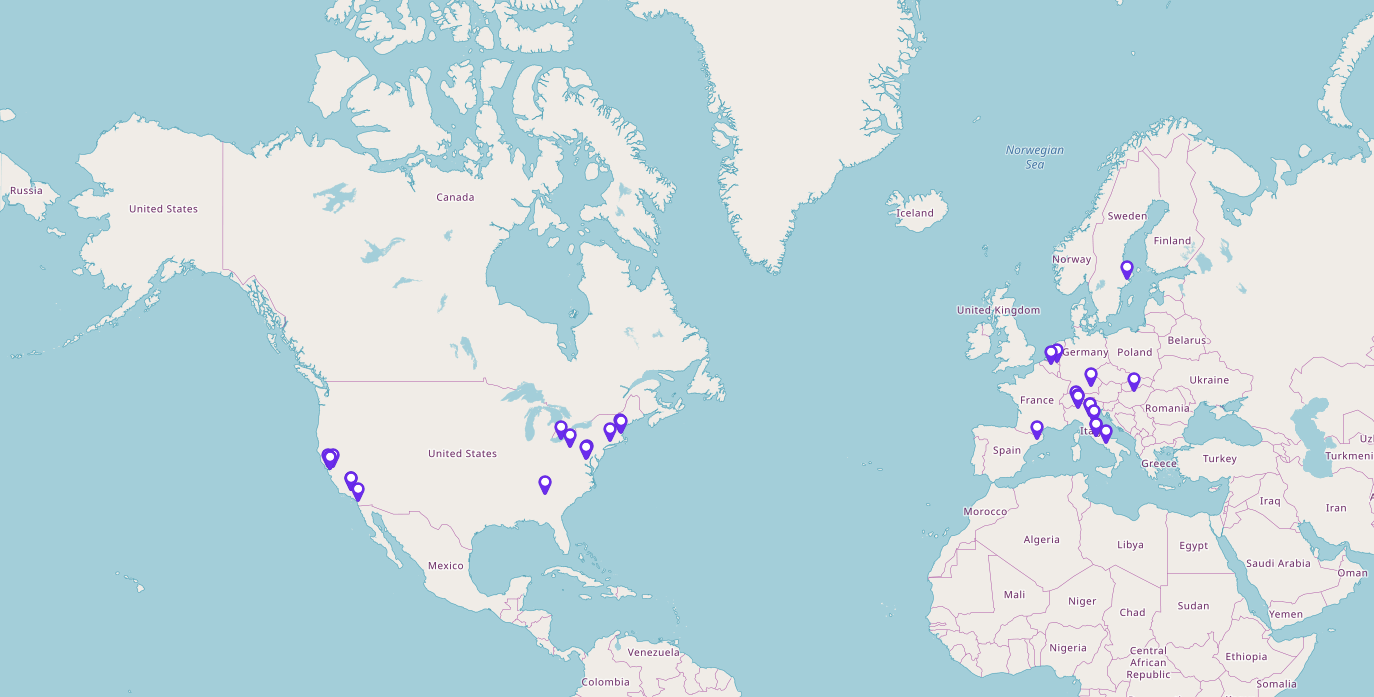
The Network Medicine Alliance represents 33 leading universities and institutions around the world. It enables members to share expertise and best practices while also providing leverage to influence health policy and clinical guidelines.
Selected Publications
















































News

Second International Conference on Network Medicine and Big Data
April 12-13, 2021
Virtual/Boston, MA
Hosted by:
Brigham and Women’s Hospital and Harvard Medical School