
A proteome-scale map of the human interactome network
Just as reference genome sequences revolutionized human genetics, reference maps of interactome networks will be critical to fully understand genotype-phenotype relationships. Here, we describe a systematic map of ∼14,000 high-quality human binary protein-protein interactions. At equal quality, this map is ∼30% larger than what is available from small-scale studies published in the literature in the last few decades. While currently available information is highly biased and only covers a relatively small portion of the proteome, our systematic map appears strikingly more homogeneous, revealing a “broader” human interactome network than currently appreciated. The map also uncovers significant interconnectivity between known and candidate cancer gene products, providing unbiased evidence for an expanded functional cancer landscape, while demonstrating how high-quality interactome models will help “connect the dots” of the genomic revolution.

Human symptoms–disease network
In the post-genomic era, the elucidation of the relationship between the molecular origins of diseases and their resulting phenotypes is a crucial task for medical research. Here, we use a large-scale biomedical literature database to construct a symptom-based human disease network and investigate the connection between clinical manifestations of diseases and their underlying molecular interactions. We find that the symptom-based similarity of two diseases correlates strongly with the number of shared genetic associations and the extent to which their associated proteins interact. Moreover, the diversity of the clinical manifestations of a disease can be related to the connectivity patterns of the underlying protein interaction network. The comprehensive, high-quality map of disease–symptom relations can further be used as a resource helping to address important questions in the field of systems medicine, for example, the identification of unexpected associations between diseases, disease etiology research or drug design.
Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins
Genotypic differences greatly influence susceptibility and resistance to disease. Understanding genotype–phenotype relationships requires that phenotypes be viewed as manifestations of network properties, rather than simply as the result of individual genomic variations. Genome sequencing efforts have identified numerous germline mutations, and large numbers of somatic genomic alterations, associated with a predisposition to cancer. However, it remains difficult to distinguish background, or ‘passenger’, cancer mutations from causal, or ‘driver’, mutations in these data sets. Human viruses intrinsically depend on their host cell during the course of infection and can elicit pathological phenotypes similar to those arising from mutations. Here we test the hypothesis that genomic variations and tumour viruses may cause cancer through related mechanisms, by systematically examining host interactome and transcriptome network perturbations caused by DNA tumour virus proteins. The resulting integrated viral perturbation data reflects rewiring of the host cell networks, and highlights pathways, such as Notch signalling and apoptosis, that go awry in cancer. We show that systematic analyses of host targets of viral proteins can identify cancer genes with a success rate on a par with their identification through functional genomics and large-scale cataloguing of tumour mutations. Together, these complementary approaches increase the specificity of cancer gene identification. Combining systems-level studies of pathogen-encoded gene products with genomic approaches will facilitate the prioritization of cancer causing driver genes to advance the understanding of the genetic basis of human cancer.

Systems biology and the future of medicine
Contemporary views of human disease are based on simple correlation between clinical syndromes and pathological analysis dating from the late 19th century. Although this approach to disease diagnosis, prognosis, and treatment has served the medical establishment and society well for many years, it has serious shortcomings for the modern era of the genomic medicine that stem from its reliance on reductionist principles of experimentation and analysis. Quantitative, holistic systems biology applied to human disease offers a unique approach for diagnosing established disease, defining disease predilection, and developing individualized (personalized) treatment strategies that can take full advantage of modern molecular pathobiology and the comprehensive data sets that are rapidly becoming available for populations and individuals. In this way, systems pathobiology offers the promise of redefining our approach to disease and the field of medicine.

Interactome Networks and Human Disease
Complex biological systems and cellular networks may underlie most genotype to phenotype relationships. Here, we review basic concepts in network biology, discussing different types of interactome networks and the insights that can come from analyzing them. We elaborate on why interactome networks are important to consider in biology, how they can be mapped and integratedwith each other, what global properties are starting to emerge from interactome network models, and how these properties may relate to human disease.

Network medicine: a network-based approach to human disease
By Albert-László Barabási, Natali Gulbahce & Joseph Loscalzo
Published: 17 December 2010

A dynamic network approach for the study of human phenotypes
The use of networks to integrate different genetic, proteomic, and metabolic datasets has been proposed as a viable path toward elucidating the origins of specific diseases. Here we introduce a new phenotypic database summarizing correlations obtained from the disease history of more than 30 million patients in a Phenotypic Disease Network (PDN). We present evidence that the structure of the PDN is relevant to the understanding of illness progression by showing that (1) patients develop diseases close in the network to those they already have; (2) the progression of disease along the links of the network is different for patients of different genders and ethnicities; (3) patients diagnosed with diseases which are more highly connected in the PDN tend to die sooner than those affected by less connected diseases; and (4) diseases that tend to be preceded by others in the PDN tend to be more connected than diseases that precede other illnesses, and are associated with higher degrees of mortality. Our findings show that disease progression can be represented and studied using network methods, offering the potential to enhance our understanding of the origin and evolution of human diseases. The dataset introduced here, released concurrently with this publication, represents the largest relational phenotypic resource publicly available to the research community.

The impact of cellular networks on disease comorbidity
The impact of disease-causing defects is often not limited to the products of a mutated gene but, thanks to interactions between the molecular components, may also affect other cellular functions, resulting in potential comorbidity effects. By combining information on cellular interactions, disease--gene associations, and population-level disease patterns extracted from Medicare data, we find statistically significant correlations between the underlying structure of cellular networks and disease comorbidity patterns in the human population. Our results indicate that such a combination of population-level data and cellular network information could help build novel hypotheses about disease mechanisms.

The implications of human metabolic network topology for disease comorbidity
Most diseases are the consequence of the breakdown of cellular processes, but the relationships among genetic/epigenetic defects, the molecular interaction networks underlying them, and the disease phenotypes remain poorly understood. To gain insights into such relationships, here we constructed a bipartite human disease association network in which nodes are diseases and two diseases are linked if mutated enzymes associated with them catalyze adjacent metabolic reactions. We find that connected disease pairs display higher correlated reaction flux rate, corresponding enzyme-encoding gene coexpression, and higher comorbidity than those that have no metabolic link between them. Furthermore, the more connected a disease is to other diseases, the higher is its prevalence and associated mortality rate. The network topology-based approach also helps to
Drug-target network
The global set of relationships between protein targets of all drugs and all disease-gene products in the human protein–protein interaction or ‘interactome’ network remains uncharacterized. We built a bipartite graph composed of US Food and Drug Administration–approved drugs and proteins linked by drug–target binary associations. The resultingnetwork connects most drugs into a highly interlinked giant component, with strong local clustering of drugs of similar types according to Anatomical Therapeutic Chemical classification. Topological analyses of this network quantitatively showed an overabundance of ‘follow-on’ drugs, that is, drugs that target already targeted proteins. By including drugs currently under investigation, we identified a trend toward more functionally diverse targets improving polypharmacology. To analyze the relationships between drug targets and disease-gene products, we measured the shortest distance between both sets of proteins in current models of the human interactome network. Significant differences in distance were found between etiological and palliative drugs. A recent trend toward more rational drug design was observed.
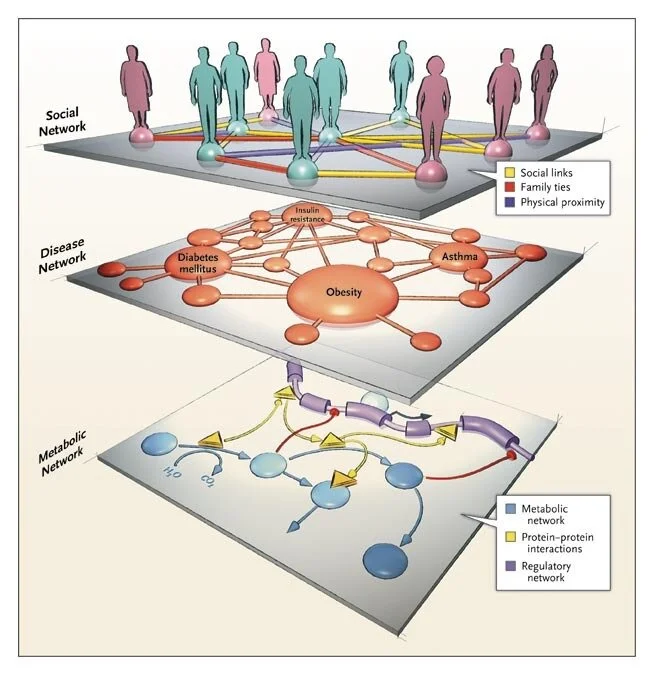
Network Medicine — From Obesity to the “Diseasome”
A recent study reported that among people who carried a single copy of the high-risk allele for the FTO gene, which is associated with fat mass and obesity, the risk of obesity increased by 30%. The risk of obesity increased by 67% among people who carried two alleles, and on average they gained 3.0 kg (6.6 lb) or more.1 Given that approximately one sixth of the population of European descent is homozygous for this allele, this link between the FTO gene and obesity appears to be one of the strongest genotype–phenotype associations detected by modern genome-screening techniques.

The human disease network
A network of disorders and disease genes linked by known disorder–gene associations offers a platform to explore in a single graphtheoretic framework all known phenotype and disease gene associations, indicating the common genetic origin of many diseases. Genes associated with similar disorders show both higher likelihood of physical interactions between their products and higher expression profiling similarity for their transcripts, supporting the existence of distinct disease-specific functional modules. We find that essential human genes are likely to encode hub proteins and are expressed widely in most tissues. This suggests that disease genes also would play a central role in the human interactome. In contrast, we find that the vast majority of disease genes are nonessential and show no tendency to encode hub proteins, and their expression pattern indicates that they are localized in the functional periphery of the network. A selection-based model explains the observed difference between essential and disease genes and also suggests that diseases caused by somatic mutations should not be peripheral, a prediction we confirm for cancer genes.

Human disease classification in the postgenomic era: A complex systems approach to human pathobiology
Contemporary classification of human disease derives from observational correlation between pathological analysis and clinical syndromes. Characterizing disease in this way established a nosology that has served clinicians well to the current time, and depends on observational skills and simple laboratory tools to define the syndromic phenotype. Yet, this time-honored diagnostic strategy has significant shortcomings that reflect both a lack of sensitivity in identifying preclinical disease, and a lack of specificity in defining disease unequivocally. In this paper, we focus on the latter limitation, viewing it as a reflection both of the different clinical presentations of many diseases (variable phenotypic expression), and of the excessive reliance on Cartesian reductionism in establishing diagnoses. The purpose of this perspective is to provide a logical basis for a new approach to classifying human disease that uses.